前言
本篇笔记主要是介绍了Bioconductor与基因组级数据分析的关系。
R语言,R包与仓库(repositories)
学习这一系列课程的前提是你已经有了一定的R语言基因。并且我们推荐使用RStudio作为R语言的使用环境。如果你跳过前面的1x-4x部分,直接进入5x,可以先参考一下其它的R语言教程,例如 Try R 。
Why R?
Bioconductor的基础就是R。使用R有3个原因:
- 许多统计学家与生物统计学家使用R语言,并且他们使用R语言设计了许多算法可以帮助我们理解复杂的实验数据。
- R语言有着高度的互操作性(interoperable),促进了使用其它语言编写软件组件的重用。
- R可移植到在各种计算机上的操作系统里(Linux、MacOSX、Windows),初学者可以使用这些平台中的任一个来学习。
总之,R的易用性和在统计学和“数据科学”中扮演着核心角色,因此当生物学家和统计学家面对基因组级的实验数据时,选择R作为处理工具就是自然而然的事情了。Bioconductor项目开始于2001年,自那以后,它就与基因组级生物学中不断增长的数据量和复杂性保持着同步。
函数式面向对象程序设计
R语言是一种混合了函数式编程与面向对象的混合型语言。
在R中进行函数式编程时,如下所示:
|
|
上面就是一个典型的函数式编程。我们定义了一个函数square(),它会计算输入的数据字的平方,即^2,例如当我们输入3时,结果就是在R中,所有的计算都是通过函数进行的。
在进行面向对象的编程时,我们所关注的生就是构建数据结构,定义能够操作这些结构化数据的方法。这是一种接近于人正常思维的编程思想。最早出现在20世纪90年代。现在我们以Bioconductor中的一个案例说明一下:
|
|
结果如下所示:
|
|
在这个案例中,我们可以看到:
OrganismDb是一个类(class);Homo.sapiens是OrganismDb这个类的一个实例(instance);- 某个类的所有方法都可以应用到这个类的所有实例中去(
methods(class=class(Homo.sapiens))就是查看这个类的方法); - 每个方法的实现是通过R中的函数进行的(我的理解就是,方法其实本质上就是函数)。并且函数的运行依赖于它自身的参数。
其中需要特别注意的是juncture(交界区)的方法,即genes,exons,transcripts,这三个方法输出基因组基本组成成分的一些信息。上面我们列出的所有方法都可以应用于OrganismDb类定义的一些实例,也就是一些模式生物,例如小鼠(Mus musculus),酒酿酵母(S. cerevisiae)和秀丽隐杆线虫(C. elegans)。
R包,模块化与持续集成(Continuous Integration)
这一部分在阅读的时候可以跳过。
R包的结构
我们可以通过编写R代码来用R执行面向对象的函数式编程。一种基本的方法就是创建“脚本”,用于定义数据的导入和分析的基本流程。当脚本以仅定义函数和数据结构的方式编写时,我们就可以将这些脚本打包,从而方便地发布,供其他有类似数据处理问题的用户使用。
R软件的打包协议规定了如何将R和其他语言的源代码与元数据以及文档一起组织起来,以促进简便的测试和发布的流程。例如,它义些文档的包的早期版本具有如下的目录和文件布局:
|
|
打包协议文档“Writing R Extensions”提供了完整的详细信息。使用R命令 R CMD build [foldername] 将对程度包文件夹的内容进行打包,从而创建可使用R CMD install[archivename] 添加到R安装的文件。不过现在都使用R Studio来完成这些任务。
模块化与包的相互依赖
打包协议可以帮助我们隔离执行有限操作集的软件,并识别随时间固有变化的程序集合的版本。没有客观的方法来确定一组操作是否适合打包。一些非常有用的包仅执行少量任务,而其他包则具有非常广泛的应用范围。重要的是打包的概念允许软件的模块化。这在两个方面很重要:范围和时间。范围的模块化对于允许并行独立开发解决不同问题的软件工具非常重要。时间上的模块化对于识别行为稳定的软件版本是很重要的。
持续集成:测试包的正确性和互操作性
下列的图片就是开发Bioconductor分支的一个构建报告的快照:

在上面的表格中,一共有6列,其中最后1列标明了“Installed Pkgs”,其中2000表示用于Linux平台,这个数据在不同平台之间有所不同,并且通常会伴随着开发分支的时间而增加。
汇总
Bioconductor的核心开发者小组致力于开发数据结构,使用户能够方便地处理基因组和基因组规模的数据。用于设计的结构是为了支持基因组规模生物学实验的主要目标:
- 解析由微阵列或测序仪产生的大规模数据集。
- 对(相对)原始数据进行预处理,以支持可靠的统计解释。
- 将分析量化与样本信息数据进行结合,用于检验分子过程和生物体水平特征(例如生长、疾病状态)之间关系的假设。
- 在本课程里,我们将回顾你可以在自己的研究中用于执行这些相关任务的无明和功能。
5x的基因前提和概述
你知道如何使用R来操作和分析数据后,并且还对统计建模有不错的理解后。Bioconductor项目就会证明,在进行基因组级规模的计算生物操作时(但非所有),R是一个有效的工具。
将Bioconductor与处理基因组数据的其它软件系统的不同之处在于:
- 使用面向对象的设计理解统一了基因组实验中出现的不同数据类型;
- 致力于基因组注释的可互操作结构,从核苷酸到人口规模;
- 发布和开发周期的持续集成原则,在多个广泛使用的计算平台上进行日常测试。我们这个为期四周的模块化学习目标旨在针对基因组规模数据分析的方方面面建立起对该系统使用的专业能力。
525.5x课程主要为了四个主要部分,每部分花费一周时间来学习,内容如下:
- 动机和技术:我们为什么要检测,以及如何检测数据,以及我们如何使用R语言来管理数据。
- 基因组注释:尤其是要注意基因组坐标中的区间(ranges,有的译为“范围”)在识别基因组结构方面的作用。
- 基因组级数据的预处理概念,重要如何通过Bioconductor来实现。
- 使用Bioconductor来做基因组级数据的假设检验。
Some of the fundamental concepts that distinguish Bioconductor from other software systems addressing genome-scale data are
本章的不同小节将简要描述一下这些概念,以及说明如何进行计算。
动机与技术
“What we measure and why”这个视频(这个视频位于Youtube上)给出了基本生物过程的示意图,我们可以通过计算来研究这些过程。我们注意到,对有机体的生命过程至关重要的所有蛋白质的编码序列都位于有机体的基因组DNA中。研究生物体之间的差异, 以及生物体内的某些变化(例如肿瘤的发展),通常涉及基因组DNA序列的计算。
Bioconductor提供了处理许多生物体基因组DNA序列的工具。一种处理序列的基本方法计算就是给定一个“参考序列”,然后我们计算另外一条序列与参考序列之间的差异。
参考序列的获取
使用Bioconductor很容易处理人类(Homo sapiens)的参考序列。现在我们来看一下17号染色体,如下所示:
|
|
我们注意到:
- 我们通过了一个R包来提供了序列。
- 包的名称表明了这个是来源于UCSC的参考基因组hg19。
- 我们使用美元符号
$提取了染色体的序列。
表示DNA突变体
与参考序列有单个偏离的标准表示方法就是使用VCF格式的文件( Variant Call Format)。VariantAnnotation包就含有这样一个案例。我们有两个某些DNA突变体的两个高质量表示方法(high-level representations),即VCF内容中的摘要,以及突变体本身在基因组上的地址,如下所示:
|
|
我们需要注意:
- 演示中的数据已经包含在了包中,它仅用于说明和测试
- 变量
vcf向用户简洁地展示了信息 - 使用
rowRanges函数提取了突变体在hg19基因组中的位置,并与标签一同显示
检测基因表达
我们将以观察模式生物酿酒酵母(Sacchomyces Cerevisiae)中基因表达的检测数值来结束这一部分的讨论。现在我们来看一个实验,这个实验检测了酿酒酵母生殖周期中一系列时间点上的全基因组mRNA丰度。我们会使用R包来管理这些数据,并且我们使用特殊的Bioconductor定义 的数据结构来访问有关的实验和结果信息,如下所示:
|
|
经过一段时间的学习,你就会在接下来的几周内成为这方面的高手,你可以将整个细胞周期中参与调控的基因随时间变化的过程绘制出来。
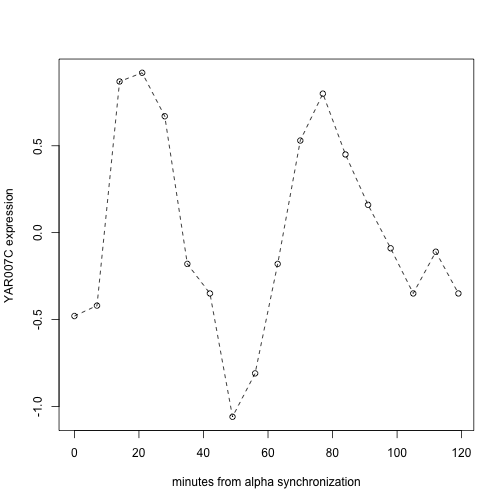
需要注意:
- 关于实验的元数据已经绑定到R包的数据中(通过
experimentData可以可看PubmedID和摘要); - 我们使用简单的语法就能选择这些复杂实验设计中的信息;在这个案例中,我们使用
spYCCES[, spYCCES$syncmeth=="alpha"]就能提取出控制alpha pheromone的基因; - R的绘图工具支持通用的绘图注释与增强功能 (plot annotation and enhancemetn);
- 统计模型工具能够直接帮我们区分周期与非周期的基因。
结束语
你即将学习一些关于基因组结构和检测它们的分子生物学技术的高水平讲座。当你遇到这些概念时,请你记住那些与理解结构以及数据处理过程相关的计算思路与方法。在Bioconductor中找到相应的计算工具,并熟练掌握这些工具。如果你找不到它们,请通知我们,或者是如果满足某些需求的工具不存在,你也可以选择构建它们。